


Теория сознания
Квантовая теория сознания - я думаю именно она ближе всего сейчас находится к истине
Квантовая теория сознания - я думаю именно она ближе всего сейчас находится к истине
Учёные МГУ сообщили о важном достижении в рамках российского квантового проекта. Им удалось масштабировать прототип квантового компьютера на нейтральных атомах рубидия до 72 кубитов. Этот результат был получен в ходе контрольного эксперимента, проводимого по дорожной карте «Росатома».

Новый вычислитель с показателем 72 кубита вошёл в тройку лидеров среди российских квантовых платформ. Ранее учёные уже продемонстрировали 70-кубитный процессор на ионах иттербия и 72-кубитный — на ионах кальция.
Особенностью нового прототипа стала обновлённая архитектура, которая разделяет вычислительный регистр на несколько зон: для хранения информации, проведения операций и считывания результатов. В эксперименте были задействованы первые две зоны. Точность выполнения двухкубитной операции составила 94%.
По словам разработчиков, следующими задачами станут дальнейшее повышение точности операций, масштабирование системы до сотен кубитов и реализация коррекции ошибок. Эти шаги приблизят создание квантового компьютера, способного решать задачи, недоступные классическим вычислительным системам.
Работа ведётся в рамках экосистемы «Росатома», объединяющей более 600 исследователей из 19 научных организаций страны.
🧠 Российские учёные создали 70-кубитный квантовый компьютер на ионах иттербия — самый мощный в стране и один из самых точных в мире. Он станет основой для применения квантовых технологий в промышленности и науке.
Трепещи крипта

Персональный квантовый компьютер, предназначенный для учебных заведений, выпустит китайская компания Shenzhen SpinQ Technology. Ориентировочная цена будущего компьютера составляет всего 5 тысяч долларов и он будет ориентирован на использование в учебных целей в школах и университетах. В прошлом году компания уже выпускала настольный квантовый компьютер по цене 50 тысяч долларов и массой 55 кг.
Новый квантовый ПК будет иметь гораздо меньшую массу, и продаваться по значительно меньшей цене. В планы компании входит выпуск серийного образца уже в четвертом квартале текущего года. Низкая стоимость компьютера, по сравнению с коммерческими квантовыми компьютерами (до 10 млн долларов) позволит приобретать устройство учебным заведениям, для которых он в первую очередь и предназначен.

Производительность квантового ПК - 2 кубита (элементарный элемент для хранения информации в квантовом компьютере), что значительно меньше существующих серийных квантовых компьютеров обрабатывающих свыше 50 кубитов. Такая невысокая производительность, однако, позволяет проводить элементарные квантовые вычисления для демонстрации принципов работы квантовых компьютеров.
Модель Gemini обладает двумя кубитами, весит 44 кг и потребляет 100 Вт. Этот компьютер способен выполнять гораздо более сложные операции. Цена вопроса составляет $43тыс. (около 3 млн руб.). Самая мощная из представленных — модель Triangulum. Ее энергопотребление составляет 300 Вт. У этого компьютера есть порт для программирования, а его квантовые схемы можно настраивать. Стоимость этой модели — $58 тыс. (более 4 млн руб.).
Базируется работа настольного квантового ПК на технологии ядерного магнитного резонанса, при котором реализуется захват молекул диметилфосфита (вещества используемого в компьютере SpinQ) мощным магнитным полем и осуществляется воздействие на них радиоволновыми импульсами, изменяющими спины отдельных атомов вещества. При этом атомы переходят в новое состояние, что аналогично переключению 1 или 0 в обыкновенных компьютерах. Изменение спина атома позволяет изменять спины соседних атомов, что создает условие для проведения математических операций.
В компьютере SpinQ используется постоянны магнит способный генерировать магнитное поле силой до Тл (тесла), что в десятки тысяч раз больше силы магнитного поля Земли. Технология была известна еще в конце прошлого века и использовалась для получения медицинских снимков. Однако тогда магнитные поля достаточной мощности могли быть созданы только с использованием сверхпроводящих магнитов, что значительно повышало стоимость устройств и увеличивало их массу и габариты. На сегодня задача решается с использованием мощных постоянных магнитов.

Малая вычислительная мощность позволяет SpinQ выполнять только несколько типичных квантовых операций, например, реализовать быстрый поиск в базе данных или версию алгоритма Гровера. Для учебных целей - демонстрации студентам основ квантовых вычислений и принципов работы квантовых компьютеров, такой мощности достаточно. Однако эти устройства никогда не сравнятся по мощности с квантовыми компьютерами, используемыми Google, IBM или Microsoft.
Сейчас будет пост про квантовые кампуктеры. Если что, я далеко не эксперт в данной области (я и с обычной-то техникой не особо дружу), и в комментариях меня можно и нужно поправлять. Сам пост написан ради вопроса в конце — если хоть кто-то понимает, как конкретно работает то, о чём я спрошу, то… Короче, я знаю, что на Пикабу обитают головастые ребята и девчата; объясните, пожалуйста, Серёже данный вопрос, мне пригодится для книжки. А ниже я распишу своё понимание.
Итак, у нас есть квантовый компьютер. Он уже существует, однако с ним крайне сложно работать, поскольку в нём от малейших внешних воздействий накапливается огромное количество ошибок, и никто не знает пока, что с этим делать. В Китае вон учёные как-то худо-бедно производят на нём вычисления, и он реально вычисляет невероятно быстро, быстрее самого мощного суперкомпьютера, работающего на обычной двухбитной системе, только вот приходится каждый раз эти вычисления за ним перепроверять, и он практически всегда работает с погрешностью.
Если совсем несведущим охота понять сам принцип работы квантового компа, то вкратце:
Вот у меня стоит дома обычный двухбитный кампуктер, его два бита — это два состояния, включено и выключено, единица и ноль, с помощью которых и производятся все вычисления в системе. Их можно производить быстрее, если нарастить мощность "железа" (воткнуть себе в системник продвинутый процессор, оперативку и т.д.), однако рано или поздно, наращивая мощность, ты уткнёшься в ограничения самого "железа" — именно поэтому все так носятся вокруг процессоров, которые становятся всё меньше и меньше размером. Проблема в том, что теперь уменьшать процессоры тупо некуда, инженеры уже чуть ли не на атомном уровне пытаются их усовершенствовать. Чтоб вы понимали, самый маленький из существующих на данный момент процессоров имеет объём 0,04 кубических миллиметра.

Здесь зашифровано "вы тупые кожаные мешки"
Однако есть другая мето́да ускорить вычисления, и называется она — Её Величество Квантовая Механика. Как всегда, поначалу вообще была голая теория — после появления самого понятия квантмеха учёным Ричарду Фейнману и Юрию Манину пришла в голову идея квантового компьютера. Позднее был написан алгоритм Шора, созданы первые прототипы, а в 2019-ом году компания Google продемонстрировала так называемое "квантовое превосходство", то есть произвела на своём квантовом компе расчёты, недоступные классическим суперкомпьютерам на двухбитной системе. Словом, всё это — не просто словоблудие, а вполне себе рабочая технология, которой занимаются в том числе в России, а конкретно в ФИАН и МФТИ.
Но как это работает?
Если я начну здесь рассказывать про квантовую механику, двухщелевой эксперимент и прочее-прочее, то получится не пост, а целый ПОСТИЩЕ, поэтому ограничусь баянистым и всем уже приевшимся котом. Только котом непростым, а котом Шрёдингера.
Надеюсь, все слыхали про этот мысленный эксперимент? Ну про блохастика в коробке, который жив и мёртв одновременно до тех пор, пока коробку не откроет сторонний наблюдатель. Слышали же, да?..

Так вот, в мысленном эксперименте Шрёдингера скрытый от наблюдателя кот может быть и жив, и мёртв, а также он может быть ОДНОВРЕМЕННО живым и мёртвым — в этот момент наш Барсик находится в так называемом состоянии суперпозиции. Точно так же, если перекладывать этот мысленный эксперимент на принцип работы компьютерных систем, то у нас может быть два бита (единица и ноль, вкл/выкл), а кубит — это то самое состояние суперпозиции, то бишь по сути дела кубит — это буквально НЕСУЩЕСТВУЮЩИЙ В НАШЕЙ РЕАЛЬНОСТИ ТРЕТИЙ БИТ ИНФОРМАЦИИ.
И откуда он вдруг, падла такая, взялся — решительно непонятно, но он, сука, есть!
А если у нас есть три бита вместо двух, то и вычисления с их помощью можно производить куда быстрее, при этом не заморачиваясь с уменьшением и улучшением "железа". Причём добавление этого третьего бита меняет скорость вычислений в геометрической прогрессии, повышает её не в два, не в три и даже не в десять, а в миллиарды раз. Даже сейчас на несовершенных квантовых компах уже производят такие операции, на которые лучшему в мире двухбитному компьютеру понадобились бы тысячи лет. Но, повторюсь, эта технология ещё очень несовершенна, поэтому их и не используют повсеместно. К тому же дорогие они…
Теперь отвлечёмся ненадолго и вернёмся к нашему живому/мёртвому Барсику в коробке. Точно так же, как на состояние суперпозиции кота влияет внешний фактор (наблюдатель) – на суперпозицию квантового компьютера аналогичным образом влияют различные факторы из внешнего мира.
Главный недостаток устройства заключается в том, что он выходит из строя и начинает генерить ошибки от любых внешних воздействий — шума, света, вибрации и т.д. Стоит незадачливому исследователю громко пёрнуть около этого чуда техники, и оно непременно выйдет из строя. И это я не шучу сейчас, действительно громким пердежом можно нарушить чистоту эксперимента и вывести наш крутой камплюктер из состояния суперпозиции. На какой-то фотографии из китайской лаборатории я видел, что инженеры ходят около компа (напоминающего, кстати, огромную люстру) в спецодежде, респираторах и мягких тапочках, причём ходят буквально на цыпочках и боятся даже чихнуть ненароком, а то вдруг там всё поломается нахрен. Иначе говоря – кот станет либо живым, либо мёртвым, и смысла от него уже не будет никакого при обоих исходах, карета превратится в тыкву, а навороченный суперкомпьютер станет просто красивой люстрой с мигающими гирляндами.

Вот такой он, квантовый комплюдахтер...
И вот знаете, я вроде бы сверху всё просто и понятно расписал, но сам, погружаясь в тему, я понимаю только одно – что я нихрена не понимаю.
Откуда вообще берётся кубит? Как его вводят в состояние квантовой запутанности? Это что, чистая математика или они как-то применяют двухщелевой эксперимент, и именно поэтому китайский Цзючжан называется фотонным компьютером? Каким образом производят эти вычисления? Условно говоря, обычный компьютер, если у тебя стоит пароль в двоичном коде, пытается решить его как-то так: 01011001, и обычный комп такой: «ага, сначала или 01, или 10, затем или 01, или 10 и т.п.» А кубитный одновременно проверит оба варианта и предложит им связку с третьим. И так далее, по нарастающей. И очень быстро. И всё благодаря квантовой запутанности, когда проходят неимоверно огромные массивы данных через женскую интуицию типа «ну не знаю, как-то так, оно само собой получилось, но ответ-то верный!»
КАК ЭТО ВООБЩЕ, СУКА, РАБОТАЕТ, ОБЪЯСНИТЕ МНЕ, ПОЖАЛУЙСТА!

Если вам понравился данный пост, то можете подписаться на аккаунт или моё сообщество в ВК, там куда больше текстов про всё на свете: Artificial Intelligence
Научная группа Физического института имени П. Н. Лебедева РАН (ФИАН) в рамках проекта под руководством Госкорпорации «Росатом» представила прототип самого мощного на сегодняшний день российского квантового компьютера. Система, построенная на ионах иттербия, достигла уровня в 70 кубитов.

В ходе контрольного эксперимента исследователи не только успешно сформировали 70-кубитный квантовый регистр, но и продемонстрировали высокую точность операций: однокубитные выполняются с точностью 99,98%, а двухкубитные — 96,1%. Это ключевой показатель, определяющий реальную вычислительную мощность системы.
Данное достижение, реализованное под руководством директора ФИАН академика Н.Н. Колачевского, является важным шагом российской дорожной карты по развитию квантовых вычислений. Оно закладывает основу для расширения круга решаемых задач и приближает создание к 2030 году квантовых компьютеров среднего масштаба, способных выполнять сложные алгоритмы для прикладных отраслей промышленности с применением коррекции ошибок.
Николай Колачевский, академик РАН, директор ФИАН: «Достижение 70-кубитного уровня квантового вычислителя говорит о том, что мы научились работать со значительным количеством кубитов. Иными словами, по количеству кубитов мы соответствуем мировой динамике (хотя ситуация постоянно развивается, и нам следует оставаться на острие). Вместе с тем мы видим своей важной задачей увеличение достоверности операций. Зарубежные компании проходили этот путь десятилетиями. Нам предстоит его пройти намного быстрее».
Илья Семериков, научный сотрудник ФИАН отметил: « Достигнутый 70-кубитный показатель – это важный этап перед переходом к планарным технологиям, с которыми связано дальнейшее увеличение мощности ионных квантовых компьютеров. К слову, в этом году в рамках дорожной карты нашей группой был продемонстрирован захват одиночных ионов в планарную ионную ловушку и проведение однокубитных операций в планарных ионных ловушках. Мы очень довольны 70-кубитным результатом, но нацелены на дальнейшую работу по развитию мощности квантовых вычислителей и их практическому применению».

Квантовый компьютер.(Courtesy)
Quantum Art : ставка на аппаратное обеспечение с амбициозным планом развития.
Последняя сделка состоялась 10 декабря когда компания Quantum Art объявила о привлечении 100 миллионов долларов в рамках раунда финансирования серии А , в результате чего общий объем привлеченных средств достиг 124 миллионов долларов. Раунд возглавила компания Bedford Ridge Capital при участии Battery Ventures, Destra Investments, Lumir Growth Partners, Disruptive AI, Harel Insurance и других, а также при продолжающихся инвестициях от Amiti Ventures, StageOne Ventures, Vertex Ventures, Entrée Capital и Института науки Вейцмана.
Компания, основанная как дочернее предприятие группы профессора Роэ Озери из Института Вейцмана, возглавляется доктором Талем Давидом (генеральный директор), доктором Амитом Бен Кишем (технический директор) и Озери (главный научный сотрудник).
Она специализируется на квантовых вычислениях с использованием захваченных ионов — области, давно известной своей точностью, но критикуемой за масштабируемость. Компания Quantum Art утверждает, что решила ключевые проблемы с помощью собственных технологий в области многокубитных вентилей, модульных архитектур и надежной коррекции ошибок.
В июне компания представила необычайно подробную дорожную карту, нацеленную на достижение цели Quantum Advantage к 2027 году и создание системы с миллионом кубитов к 2033 году. План включает в себя систему с 50 кубитами в следующем году; линейку «Perspective» с 1000 кубитами в 2027 году; сверхплотную платформу «Landscape» с 12 000–40 000 кубитами; и, наконец, отказоустойчивую архитектуру «Mosaic».
Classiq: Программное обеспечение как недостающий слой
Что касается программного обеспечения, в ноябре Classiq привлекла около 30 миллионов долларов в рамках дополнительного раунда финансирования, в котором также приняли участие AMD Ventures, Qualcomm Ventures, IonQ и крупные финансовые институты, такие как Mirae Asset Capital, LeumiTech77 от Bank Leumi и Quantum Eretz. На сегодняшний день компания привлекла более 200 миллионов долларов, после завершения раунда финансирования серии C на сумму 110 миллионов долларов всего шестью месяцами ранее и дополнительных инвестиций в размере 10 миллионов долларов от SoftBank.

Основатели Classiq.( Фото: Эяль Туэг )
Компания Classiq создает операционную систему и среду разработки, которые преобразуют высокоуровневые задачи в квантовые схемы, позволяя организациям создавать приложения без глубоких знаний в области квантовой физики. Партнерские отношения с NVIDIA, Microsoft и AWS, а также с такими клиентами, как BMW Group, Comcast, Rolls-Royce, Citi, Toshiba и SoftBank, свидетельствуют о том, что предприятия все чаще видят ценность в подготовке к квантовым вычислениям за несколько лет до того, как оборудование достигнет зрелости.
Компания, основанная в 2020 году генеральным директором Ниром Минерби, директором по продуктам Амиром Навехом и техническим директором доктором Йехудой Навехом, насчитывает 100 сотрудников, три четверти из которых работают в Израиле.
QuamCore: Гонка за миллионом кубитов
В августе компания QuamCore привлекла 26 миллионов долларов в рамках раунда финансирования серии А , в результате чего общий объем привлеченных средств достиг 35 миллионов долларов, включая грант в размере 4 миллионов долларов от Управления инноваций Израиля. Раунд возглавила компания Sentinel Global при участии Arkin Capital и уже привлеченных инвесторов Viola Ventures, Earth & Beyond Ventures, Surround Ventures, Rhodium и Qbeat.

Основатели QuamCore.( Фото: QuamCore )
Компания QuamCore утверждает, что разработала полностью спроектированную и смоделированную архитектуру для масштабирования сверхпроводящих квантовых систем до одного миллиона кубитов в одном криостате, что значительно превосходит предел в ~5000 кубитов на модуль, достигнутый Google и IBM.
В случае подтверждения эффективности этот подход коренным образом перепишет представления о физических пределах сверхпроводящих систем.
Компанию возглавляют генеральный директор Алон Коэн, ранее работавший в группе EyeC Radar компании Mobileye, а также технический директор профессор Шей Хакоэн-Гуржи и главный научный сотрудник профессор Серж Розенблюм, оба ведущие специалисты в области сверхпроводящих квантовых исследований в Технионе и Институте Вейцмана. Их совместные научные работы были опубликованы в журналах Science, Nature и других ведущих изданиях.
Qedma: Решение самой большой проблемы квантовых вычислений
Частота ошибок остается определяющим препятствием для практического применения квантовых вычислений, и израильский стартап Qedma занял именно эту узкую точку. В июле компания привлекла 26 миллионов долларов в рамках раунда финансирования серии А, возглавляемого Glilot+, при участии IBM, Korean Investment Partners и других инвесторов.

Команда Qedma.( Фото: Эяль Туэг )
Компания Qedma разрабатывает программное обеспечение, которое идентифицирует и изучает профиль шума каждого квантового устройства и корректирует алгоритмы для подавления и уменьшения ошибок. Компания утверждает, что ее методы позволяют проводить квантовые вычисления, в 1000 раз превышающие возможности современного оборудования. Это значительно снизит накладные расходы, необходимые для квантовой коррекции ошибок, которая обычно требует до 1000 физических кубитов на каждый логический кубит.
История компании началась в 2020 году с разговора между профессором Нетанелем Линднером и доктором Асифом Синаем, к которым позже присоединилась профессор Дорит Ахаронов, пионер теоремы об отказоустойчивости, доказавшей теоретическую возможность крупномасштабных квантовых вычислений. Их еженедельные дискуссии переросли в стартап, целью которого является создание «операционного уровня», которого в настоящее время не хватает квантовым машинам.
Quantum Machines: системы управления приобретают стратегическое значение.
Крупнейшее в этом году привлечение инвестиций произошло в феврале, когда компания Quantum Machines завершила раунд финансирования серии C на сумму 170 миллионов долларов , увеличив общий объем инвестиций до 280 миллионов долларов и оценив компанию примерно в 700 миллионов долларов. Раунд возглавила компания PSG Equity при участии Red Dot Capital Partners, Intel Capital, TLV Partners, Battery Ventures и предпринимателя Авигдора Вилленца.

Команда «Quantum Machines».( Фото: Илья Мельников )
Компания Quantum Machines разрабатывает гибридные системы управления, используемые практически во всех типах квантового оборудования. Ее технологии получили широкое распространение по всему миру, в том числе благодаря стратегическому сотрудничеству с NVIDIA в рамках проекта DGX Quantum, который объединяет квантовое управление в реальном времени с высокоскоростными классическими вычислениями.
Компания была основана в 2018 году доктором Итамаром Сиваном (генеральный директор), доктором Йонатаном Коэном (технический директор) и доктором Ниссимом Офеком (вице-президент по исследованиям и разработкам), которые являются выпускниками Субмикронного центра Института Вейцмана.
Читай по теме:
Перевод с английского
Автор: Денис Аветисян
Новый подход с использованием обучения с подкреплением позволяет автоматически проектировать квантовые схемы для эффективного вычисления энергии молекул.
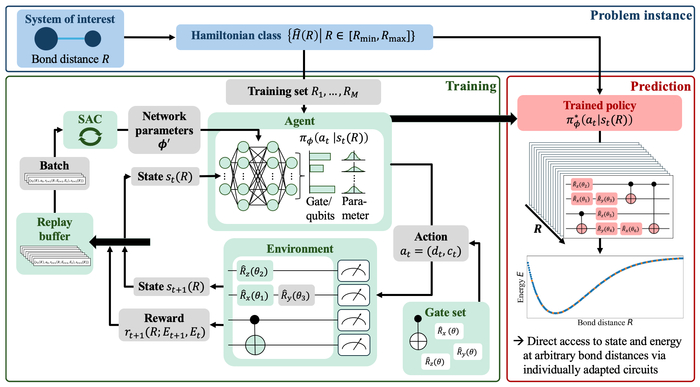
Изучается энергия основного состояния молекулярной системы как функция расстояния между атомами, при этом алгоритм обучения с подкреплением конструирует квантовые схемы с нуля, выбирая гейты и параметры, чтобы предсказывать индивидуально адаптированные схемы для произвольных расстояний и, таким образом, напрямую получать потенциальную энергию и соответствующие волновые функции.
В статье представлена платформа обучения с подкреплением для разработки переносимых квантовых схем, способных эффективно исследовать поверхности потенциальной энергии молекул.
Построение эффективных квантовых схем для моделирования сложных молекулярных систем остается сложной задачей, требующей значительных вычислительных ресурсов. В данной работе, 'Reinforcement learning of quantum circuit architectures for molecular potential energy curves', предложен подход, основанный на обучении с подкреплением, для автоматического проектирования квантовых схем, адаптированных к конкретным молекулам и их потенциальным энергетическим кривым. Разработанная методика позволяет создавать переносимые схемы, способные эффективно исследовать энергетические поверхности, что открывает путь к масштабируемым квантовым симуляциям. Сможет ли данный подход значительно ускорить разработку новых материалов и лекарственных препаратов благодаря более точным квантово-химическим расчетам?
Традиционное молекулярное моделирование сталкивается с существенными вычислительными ограничениями, препятствующими детальному изучению больших и сложных систем. Вычисление энергии основного состояния, определяющей ключевые характеристики молекулярного поведения, часто становится непосильной задачей при увеличении размеров моделируемой молекулы. В этой связи, квантовые вычисления предлагают принципиально новый подход к решению проблемы, однако для реализации этого потенциала необходима разработка эффективных алгоритмов и специализированных квантовых схем, способных оптимально использовать возможности квантовых систем для моделирования молекулярных процессов и предсказания их свойств.
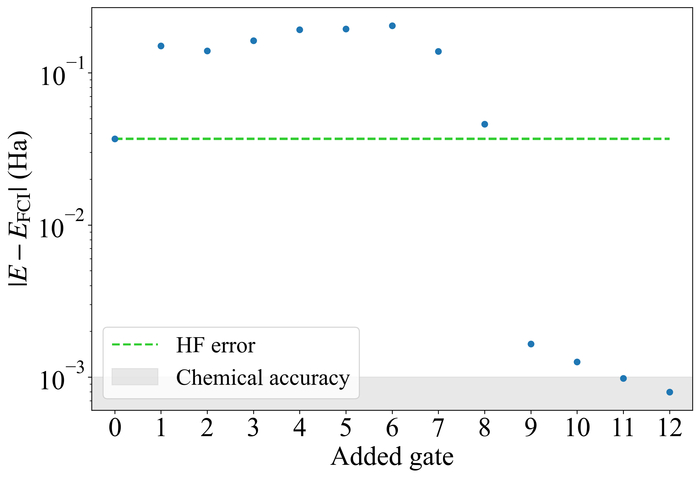
Обученная схема демонстрирует снижение ошибки энергии относительно энергии FCI с увеличением числа вентилей для молекулы LiH, состоящей из четырех кубитов.
Разработанный подход использует принципы усиленного обучения для автоматизированного проектирования квантовых схем, предназначенных для вычисления энергий молекул. Агент, функционирующий в рамках данной системы, взаимодействует с гибридной квантово-классической симуляцией, в качестве функции оценки используя вариационный квантовый решатель собственных значений (VQE). Этот итеративный процесс позволяет агенту последовательно оптимизировать структуру и параметры квантовой схемы, стремясь к минимизации рассчитанной энергии и достижению более точного представления молекулярной системы. В результате, предложенный метод демонстрирует пятикратное (5.1x) увеличение точности по сравнению с приближением Хартри-Фока.
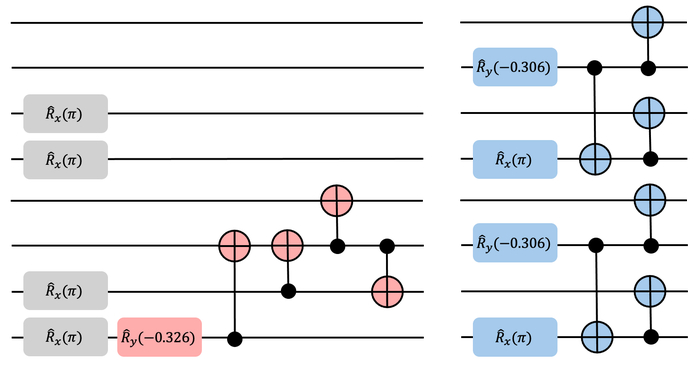
Обученная модель обучения с подкреплением (красный) генерирует структуру, эффективно запутывающую кубиты четыре через семь на всем диапазоне межатомных расстояний, в отличие от стандартного SPA-анзаца (синий), состоящего из двух отдельных блоков запутывания.
Для обучения агента использовался алгоритм Soft Actor-Critic (SAC), эффективно балансирующий исследование и использование полученного опыта. Ключевым элементом является регуляризация энтропии, побуждающая к изучению разнообразных структур и параметров квантовых схем, что предотвращает преждевременную сходимость к субоптимальным решениям. Стабилизация процесса обучения достигается благодаря использованию Target Network, обеспечивающего согласованную и отложенную оценку ценности. Алгоритм успешно справляется как с дискретными, так и с непрерывными пространствами действий, позволяя осуществлять точный контроль над проектированием квантовых схем. Применительно к молекуле LiH, состоящей из шести кубитов, удалось достичь средней ошибки в 0.0161 Ха с отклонением +0.0136/-0.0036 Ха.
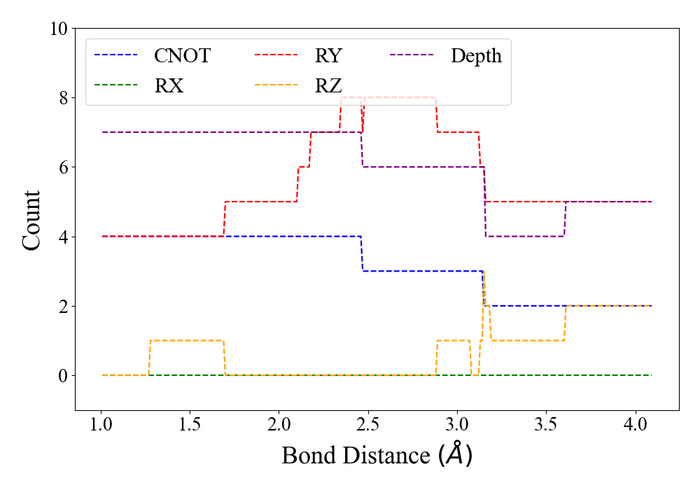
Анализ количества операций CNOT, Rx, Ry, Rz и общей глубины схемы для молекулы LiH из четырех кубитов в диапазоне межатомных расстояний от 1.0 до 4.0 Å показывает зависимость этих параметров от геометрии молекулы.
Для повышения эффективности обучения агента применяется буфер повторного использования опыта, в котором сохраняются данные о его взаимодействиях со средой - состояние, действие, полученное вознаграждение и следующее состояние. Такой подход позволяет эффективно использовать накопленные данные, устраняя корреляции между последовательными выборками и позволяя агенту учиться на прошлых успехах и неудачах, что значительно ускоряет сходимость процесса обучения. В контексте оптимизации квантовых схем, данная методика существенно снижает вычислительные затраты и время, необходимое для достижения оптимального решения, что делает возможной работу с более крупными и сложными молекулярными системами. В частности, применение данного подхода позволило снизить стоимость обучения в 22 раза по сравнению с оптимизацией на фиксированном расстоянии, при этом средняя ошибка для молекулы LiH, состоящей из четырех кубитов, составила 0.0136 с отклонением +0.0156/-0.0096 Ha.
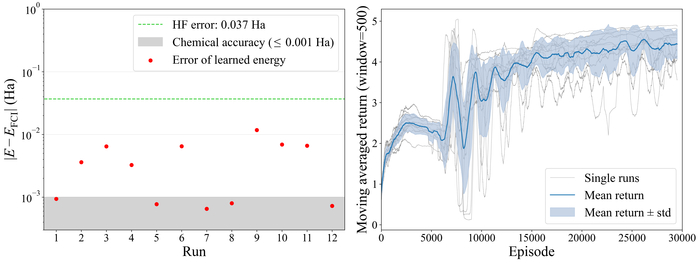
Обучение на четырехкубитном LiH демонстрирует сходимость энергии и стабильный возврат в каждой из двенадцати итераций.
Исследование, представленное в данной работе, демонстрирует, что создание эффективных квантовых схем для моделирования молекулярных систем требует не жесткого планирования, а скорее адаптации и эволюции. Подобно тому, как экосистема формируется естественным отбором, квантовые схемы, разработанные с использованием обучения с подкреплением, демонстрируют способность к переносу знаний и эффективному исследованию поверхностей потенциальной энергии. Как говорил Альберт Эйнштейн: «Фантазия важнее знания. Знание ограничено. Фантазия охватывает весь мир». Этот принцип применим и здесь: алгоритм не просто ищет оптимальное решение, а создает основу для дальнейшего развития и адаптации, предвидя и смягчая будущие сбои в сложных молекулярных системах. Архитектура, предложенная авторами, - это не инструмент, а экосистема, способная к самоорганизации и выживанию.
Представленная работа, стремясь к автоматизированному проектированию квантовых схем, неизбежно сталкивается с фундаментальной дилеммой. Каждая оптимизация, каждое «обучение с подкреплением» - это лишь временное усмирение хаоса, запрограммированное проявление будущей хрупкости. Схемы, кажущиеся эффективными сегодня, несут в себе семена собственной деградации, проявляющиеся в новых молекулярных ландшафтах, с которыми они не были обучены. В каждом кроне этой «эволюции» скрыт страх перед неожиданным, перед тем, что не вписывается в узкие рамки текущего обучения.
Надежда на «идеальную» квантовую архитектуру, способную охватить всю сложность потенциальных поверхностей, - это форма отрицания энтропии. Более вероятен путь постепенного накопления «шрамов» - ограниченных, но устойчивых решений для конкретных классов молекул. Истинный прогресс, вероятно, лежит не в создании универсальной схемы, а в разработке механизмов быстрого восстановления после неизбежных сбоев, в адаптации к новым вызовам, а не в их предвидении.
Перспективы переноса обучения, намеченные в данной работе, кажутся особенно тревожными. Каждый перенос - это риск привнести скрытые зависимости, которые проявятся в неожиданный момент, превратив полезный инструмент в источник систематических ошибок. В конечном итоге, успех этого направления будет зависеть не от скорости обучения, а от способности к самодиагностике и отказу от ошибочных стратегий.
Полный обзор с формулами: denisavetisyan.com/kvantovye-shemy-uchatsya-modelirovat-molekuly
Оригинал статьи: https://arxiv.org/pdf/2511.16559.pdf
Связаться с автором: linkedin.com/in/avetisyan






